
RNA-Seq

A RNA-Seq é unha técnica de bioloxía molecular que utiliza secuenciación de seguinte xeración para revelar a presenza e cantidade de ARN nunha mostra biolóxica nun momento dado, analizando o transcritoma celular en constante cambio.[2][3]
Especificamente, a RNA-Seq facilita a capacidade de observar transcritos de xenes de empalme alternativo, modificacións postranscricionais, fusións de xenes, mutacións/SNPs e cambios na expresión xénica co tempo ou diferenzas en expresión xénica entre diferentes grupos ou tratamentos.[4] Ademais dos transcritos e ARNm, a RNA-Seq pode analizar diferentes poboacións de ARN, incluíndo o ARN total, o ARN pequeno, como o miARN, o ARNt e o perfil ribosómico.[5] A RNA-Seq pode tamén ser usado para determinar as fronteiras exón/intrón e verificar ou emendar os límites 3' e 5' anotados previamente. Os avances recentes en RNA-Seq inclúen a secuenciación de célula única e a secuenciación in situ de tecidos fixados.[6]
Antes da aparición da RNA-Seq, os estudos de expresión xénica facíanse con hibridacións baseadas en micromatrices. Algúns problemas que tiñan as micromatrices son: artefactos de hibridación cruzada, deficiente cuantificación de xenes que se expresan pouco ou moito e necesidade de coñecer a secuencia a priori.[7] Debido a estas técnicas, a transcritómica fixo a transición a métodos baseados na secuenciación. Estes progresaron desde a secuenciación de Sanger de bibliotecas de marcadores de secuencia expresada (ETS, Expressed Sequence Tag), a métodos baseados en etiquetas químicas (por exemplo, a análise en serie da expresión xénica) e finalmante á tecnoloxía actual, a secuenciación de xene seguinte de ADNc (notablemente a RNA-Seq).
Métodos
[editar | editar a fonte]Preparación de bibliotecas
[editar | editar a fonte]- Véxase tamén: Biblioteca (bioloxía).

Os pasos xerais que hai que seguir para preparar unha biblioteca de ADN complementario (ADNc) para secuenciar descríbense máis abaixo, pero adoitan variar entre plataformas, e son:[3][8][9]
- Illamento do ARN: o ARN é illado dun tecido e mesturado con desoxirribonuclease (DNase). A DNase reduce a cantidade de ADN xenómico. O grao de degradación do ARN compróbase con electroforese en xel e electroforese de capilaridade e é utilizado para asignar un número de integridade do ARN á mostra. Esta calidade do ARN e a cantidade total de ARN do comezo téñense en consideración durante os subseguintes pasos de preparación da biblioteca, secuenciación e análise.
- Selección/depleción do ARN: para analizar os sinais de interese, o ARN illado pode ser mantido como tal, filtrado para obter ARN con colas 3' poliadeniladas (poli(A)) para así incluír só os ARNm, eliminándose o ARNr), e/ou filtrado para ARN que se una a secuencias específicas (ver métodos de selección e depleción de ADN na táboa de máis abaixo). Os ARN con colas 3' poli(A) son secuencias codificantes procesadas maduras. A selección de poli(A) realízane mesturando ARN con oligómeros poli(T) unidos covalentemente a un substrato, normalmente boliñas magnéticas.[10][11] A selección de poli(A) ignora o ARN non codificante e introduce un nesgo 3',[12] o cal se evita coa estratexia de depleción de ARN ribosómico. O ARNr é eliminado porque supón un 90% do ARN da célula, que se se mantivese daría lugar a outros datos no transcritoma.
- Síntese de ADNc: o ARN é reversotranscrito a ADNc porque o ADN é máis estable e permite a amplificación (a cal usa ADN polimerases) e facer uso da tecnoloxía de secuenciación de ADN máis maduro. A amplificación que segue á transcrición inversa ten como resultado a perda da direccionalidade ou sentido da febra (strandedness), o cal se pode evitar co etiquetado químico ou secuenciación de molécula única. Realízanse a fragmentación e selección de tamaños para purificar secuencias que son da lonxitude axeitada para a máquina de secuenciación. O ARN, o ADNc, ou ambos son fragmentados con enzimas, por sonicación ou con nebulizadores. A fragmentación do ARN reduce o nesgo 5' da transcrición inversa cebada aleatoriamente e a influencia de sitios de unión ao cebador,[11] co inconveniente de que os extemos 5' e 3' son convertidos en ADN menos eficientemente. Despois da fragmentación faise a selección por tamaños, na que ou ben se eliminan pequenas secuencias ou ben é seleccionado un estreito rango de lonxitudes de secuencias. Como se perden os pequenos ARNs como os microARNs, estes son analizados independentemente. O ADNc para cada experimento pode ser indexado cun código de barras hexámero ou octámero, para que estes experimentos poidan poñerse nun só carreiro de secuenciación multiplexada.
| Estratexia | Tipo de ARN | Contido en ARN ribosómico | Contido en ARN non procesado | Contido en ADN xenómico | Método de illamento |
|---|---|---|---|---|---|
| ARN total | Todo | Alto | Alto | Alto | Ningún |
| Selección de poliA | Codificante | Baixo | Baixo | Baixo | Hibridación con oligómeros poli(dT) |
| Depleción de ARNr | Codificante, non codificante | Baixo | Alto | Alto | Eliminación de oligómeros complementarios do ARNr |
| Captura de ARN | Dirixido a diana | Baixo | Moderado | Baixo | Hibridción con sondas complementarias a transcritos desexados |
Secuenciación de ARN pequeno/ARN non codiicante
[editar | editar a fonte]Cando se secuencian outros ARNs que non son ARNm, a biblioteca é modificada. O ARN celular é seleccionado baseándose no rango de tamaños desexado. Para dianas de ARN pequeno, como o miRNA, o ARN íllase por selección de tamaños. Isto pode ser realizado cun xel de exclusión por tamaños, por medio de boliñas magnéticas de selección de tamaños ou cun kit desenvolvido comercialmente. Unha vez illado, engádense linkers aos extremos 3' e 5' e despois son purificados. O paso final é a xeración de ADNc por transcrición inversa.
Secuenciación de ARN directa
[editar | editar a fonte]

Como se viu que a conversión de ARN en ADNc, a ligación, amplificación e outras manipulacións da mostra introducen nesgos e artefactos que poden interferir tanto coa propia caracterización e a cuantificación de transcritos,[13] A secuenciación de ARN directa de molécula única foi explorada por compañías como Helicos (que xa cerrou), Oxford Nanopore Technologies,[14] e outras. Esta tecnoloxía secuencia as moléculas de ARN directamente de maneira masivamente paralela.
Secuenciación de ARN de célula única (scRNA-Seq)
[editar | editar a fonte]- Véxase tamén: Secuenciación de célula única.
Os métodos estándar como as micromatrices e a análise RNA-Seq en masa estándar analizan a expresión de ARNs a partir de grandes poboacións de células. En poboacións de células mesturadas, estas medidas poden ocultar diferenzas esenciais entre células individuais nesas poboacións.[15][16]
A secuenciación de ARN de célula única ou scRNA-Seq (do inglés single-cell RNA sequencing) proporciona os perfís de expresión de células individuais. Aínda que non é posible obter a información completa de todo o ARN expresado por cada célula, debido á pequena cantidade de material dispoñible, os padróns de expresión xénica poden ser identificados por análises de clústeres de xenes. Isto pode desvelar a existencia de tipos raros de células dentro dunha poboación de células que nunca puideron observarse antes. Por exemplo, en 2018 foron identificados células especializadas raras do pulmón chamadas ionocitos pulmonares que expresan o regulador de condutancia de membrana da fibrose quística por dos grupos que realizaron unha scRNA-Seq dos epitelios das vías aéreas plmonares.[17][18]
Procedementos experimentais
[editar | editar a fonte]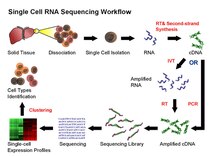
Os protocolos actuais de scRNA-Seq comprenden os seguintes pasos: o illamento de célula única e ARN, transcrición inversa, amplificación, xeración de bibliotecas e secuenciación. Os métodos que apareceron inicialmente separaban as células individuais en pozos separados; os métodos máis recentes encapsulan as células individuais en pingas nun aparello microfluídico, onde ten lugar a reacción da transcrición inversa, convertendo os ARNs en ADNc. Cada pinga leva un "código de barras" de ADN que etiqueta unicamente o ADNc derivado dunha soa célula. Unha vez que se completa a transcrición inversa, pode mesturarse todo o ADNc de moitas células para a secuenciación; os transcritos dunha célula determinada identifícanse polo código de barras exclusivo.[19][20]
Os retos aos que se enfronta a scRNA-Seq inclúen a conservación da abundancia inicial de ARNm nunha célula e a identificación de transcritos raros.[21] A etapa da transcrición inversa é fundamental, xa que a eficiencia da reacción de reversotranscrición (RT) determina canta poboación de ARN da célula será finalmente analizado polo secuenciador. A procesividade das transcriptases inversas e as estratexias de cebado usadas poden afectar á produción de ADNc de lonxitude completa e a xeración de bibliotecas nesgadas cara aos extremos 3’ ou 5' dos xenes.
No paso de amplificación, úsanse actualmetne tanto a PCR coma a transcrición in vitro (IVT) para amplificar o ADNc. Unha das vantaxes de métodos baseados na PCR é a capacidade de xerar ADNc de lonxitude completa. Porén, as diferentes eficiencias da PCR sobre secuencias determinadas (por exemplo, o contido GC) poden tamén ser amplificadas exponencialmente, producindo bibliotecas con cobetura desigual. Por outra parte, mentres que as bibliotecas xeradas por IVT poden evitar o nesgo de secuencias inducido pola PCR, unha secuencia específica pode ser transcrita ineficazmente, causando así a desaparición (drop-out) de secuencias ou a xeración de secuencias incompletas.[15][22] Publicáronse varios protocolos para a scRNA-Seq: Tang et al.,[23] STRT,[24] SMART-seq,[25] CEL-seq,[26] RAGE-seq,[27] , Quartz-seq.[28] e C1-CAGE.[29] Estes protocolos diferéncianse nas súas estratexias para a transcrición inversa, síntese de ADNc e a amplificación, e a posibilidade de aloxar códigos de barra específicos de secuencia (é dicir, UMIs) ou a capacidade de procesar mostras combinadas.[30]
En 2017, introducíronse dúas estratexias para medir simultaneamente o ARNm e expresión proteica de célula única por medio de anticorpos etiquetados con oligonucleótidos coñecidas como REAP-seq,[31] e CITE-seq.[32]
Aplicacións
[editar | editar a fonte]A scRNA-Seq está sendo amplamente usada en disciplinas biolóxicas como a bioloxía do desenvolvemento, neuroloxía,[33] oncoloxía,[34][35][36] enfermidades autoinmunes,[37] e doenzas infecciosas.[38]
A scRNA-Seq propiciou moitos avances na comprensión do desenvolvemento de embrións e organismos, como o verme Caenorhabditis elegans,[39] e a planaria rexenerativa Schmidtea mediterranea.[40][41] Os primeiros vertebrados que foron mapados desta maneira foron o peixe cebra[42][43] e o anfibio Xenopus laevis.[44] En cada caso foron estudados múltiples estadios do desenvolvemento do embrión, o que permite que o proceso de desenvolvemento completo sexa mapado célula por célula.[8] A revista Science recoñeceu estes avances como o Logro do ano 2018.[45]
Consideracións experimentais
[editar | editar a fonte]Cando se deseñan e realizan experimentos de RNA-Seq considéranse unha serie de parámetros:
- Especificidade de tecido: a expresión xénica varía dentro dun tecido e entre diferentes tecidos, e a RNA-Seq mide esta mestura de tipos de células. Isto pode facer difícil illar o mecanismo biolóxico de interese. A secuenciación de célula única pode utilizarse para estudar cada célula individualmente, o que atenúa este problema.
- Dependencia do tempo: a expresión xénica cambia co tempo e o que fai a RNA-Seq soamente é sacar unha instantánea. Poden realizarse experimentos que teñen en conta o decurso do tempo para observar cambios no transcritoma.
- Cobertura (tamén chamada profundidade): o ARN contén as mesmas mutacións observadas no ADN e a detección require unha cobertura máis profunda. Cunha cobertura suficientemente alta, a RNA-Seq pode utilizarse para estimar a expresión de cada alelo. Isto pode roporcionar coñecementos sobre fenómenos como a impresión xenética ou os efectos regulatorios en cis. A profundidade de secunciación que cómpre para aplicacións específicas pode ser extrapolada dun experimento piloto.[46]
- Artefactos de xeración de datos (tamén chamados varianza técnica): os reactivos (por exemplo, o kit de preparación de bibliotecas), persoal implicado e tipo de secuenciador (por exemplo, Illumina, Pacific Biosciences) poden orixinar artefactos técnicos que poderían ser mal interpretados como resultados significativos. Como ocore con calquera experimento científico, é prudente realizar a RNA-Seq nunha instalación ben controlada. Se isto non é posible ou o estudo é unha metaanálise, outra solución é detectar artefactos técnicos por inferencia de variables latentes (tipicamente a análise de compoñentes principais ou análise de factores) e seguidamente corrixir estas variables.[47]
- Xestión de datos: un só experimento de RNA-Seq en humanos é xeralmente da orde de 1 Gb.[48] Este gran volume de datos pode presentar problemas de almacenamento. Unha solución é comprimir os datos usando esquemas computacionais multiuso (por exemplo, gzip) ou esquemas específicos de xenómica. Estes últimos poden estar baseados en secuencias de referencia ou secuencias de novo. Outra solución é realizar experimentos de micromatrices, que poden ser dabondo para un traballo baseado nunha hipótese ou estudos de replicación (opostos á investigación exploratoria).
Análises
[editar | editar a fonte]
Ensamblaxe do transcritoma
[editar | editar a fonte]Utilízanse dous métodos para asignar lecturas de secuencia brutas a características xenómicas (é dicir, ensamblar o transcritoma):
- De novo: esta estratexia non require un xenoma de referencia para reconstruír o transcritoma e úsase normalmente cando o xenoma é descoñecido, incompleto ou está alterado substancialmente en comparación coa referencia.[49] As dificultades que se poden presentar cando se usan lecturas curtas para a ensamblaxe de novo son: 1) determinar que lecturas deberían unirse en secuencias contiguas (cóntigos), 2) a robustez para secuenciar erros e outros artefactos, e 3) a eficiencia computacional. O principal algoritmo utilizado para a ensamblaxe de novo fixo a transición desde gráficos de solapamento, que identifican todos os solapamentos por pares entre lecturas, a gráficos de de Bruijin, que rompen as lecturas en secuencias de lonxitude k e colapsan todos os k-meros nunha táboa hash.[50] Os gráficos de solapamento eran usados coa secuenciación de Sanger, pero non escalan ben os millóns de lecturas xeradas con RNA-Seq. Exemplos de ensambladores que usan gráficos de de Bruijn son Velvet,[51] Trinity,[49] Oases,[52] e Bridger.[53] A secuenciación de lecturas longas e de extremos apareados da mesma mostra pode mitigar os déficits na secuenciación de lecturas curtas o servir como molde ou esqueleto. Entre as métricas para estimar a calidade dunha ensamblaxe de novo están a lonxitude de cóntigo media, o número de cóntigos e a N50.[54]

- Guiado polo xenoma: esta estratexia baséase nos mesmos métodos utilizados para o aliñamento de ADN, coa complexidade adicional de aliñar lecturas que cobren porcións non continuas do xenoma de referencia.[55] Estas lecturas non continuas son o resultado de secuenciar transcritos empalmados (ver figura). Tipicamente, os algoritmos de aliñamento constan de dous pasos: 1) aliñar porcións curtas da lectura (é dicir, sementar o xenoma), e 2) usar programación dinámica para encontrar un aliñamento óptimo, ás veces en combinación con anotacións coñecidas. Entre as ferramentas de software que usan o aliñamento guiado polo xenoma están Bowtie,[56] TopHat (que se basea en resultados de BowTie para aliñar unións de empalme),[57][58] Subread,[59] STAR,[55] HISAT2,[60] Sailfish,[61] Kallisto,[62] e GMAP.[63] A calidade dunha ensamblaxe guiada polo xenoma pode medirse tanto con métricas de ensamblaxe de novo (por exemplo, N50) coma con comparacións para coñecer secuencias de transcritoe, unióne de empalme, xenomas e proteínas usando precisión e exhaustividade (precision and recall), ou as súas combinacións (por exemplo, o valor F1).[54] Ademais, pode realizarse a estimación in silico usando lecturas simuladas.[64][65]
Unha nota sobre a calidade da ensamblaxe: o consenso actual é que: 1) a calidade de ensamblaxe pode variar dependendo da métrica que se utilizou, 2) as ensamblaxes que teñen bos valores nunha especie non necesariamente cadran ben noutras especies, e 3) combinar diferentes estratexias podería ser o máis fiable.[66][67]
Cuantificación da expresión xénica
[editar | editar a fonte]A expresión cuanifícase para estudar os cambios celulares en resposta a estímulos externos, as diferenzas entre os estados de saúde e enfermidade, e outras cuestións a investigar. A expresión xénica utilízase a miúdo como un equivalente da abundancia de proteínas, pero estes a miúdo non son equivalentes debido a eventos postranscricionais como a interferencia de ARN e a degradación do ARNm mediada por unha mutación sen sentido.[68]
A expresión é cuantificada contando o número de lecturas que se maparon en cada locus no paso de ensamblaxe do transcritoma. A expresión pode ser cuantificada para os exóns ou xenes utilizando cóntigos ou anotacións de transcritos de referencia.[8] Estes recontos de lecturas de RNA-Seq observadas foron solidamente validadas respecto a tecnoloxías máis vellas, incluíndo as micromatrices de expresión e a qPCR.[46][69] Exemplos de ferramentas que cuantifican os recontos son HTSeq,[70] FeatureCounts,[71] Rcount,[72] maxcounts,[73] FIXSEQ,[74] e Cuffquant. Os recontos de lecturas son despois convertidos en métricas apropiadas para a comprobación de hipóteses, regresións e outras análises. Os parámetros para esta conversión son:
- Profundidade/cobertura da secuenciación: aínda que a profundidade está pre-especificada cando se realizan experimentos de RNA-Seq múltiples, esta seguirá variando moito entre un experimento e outro.[75] Por tanto, o número total de lecturas xerado nun só experimento está normalmente normalizado ao converter recontos en fragmentos, lecturas ou recontos por millón de lecturas mapadas (FPM, RPM ou CPM). A profundidade de secunciación denomínase ás veces tamaño de biblioteca, o número de moléculas de ADNc intermediario no experimento.
- Lonxitude do xene: os xenes máis longos terán máis fragmentos/lecturas/recontos que os máis curtos se a expresión de transcritos é a mesma. Isto é axustado dividindo a FPM pola lonxitude dun xene, o que ten como resultado os fragmentos métricos por quilobase de transcrito por millón de lecturas mapadas (FPKM).[76] Cando se examinan grupos de xenes en mostras, o FPKM é convertido a transcritos por millón (TPM) dividindo cada FPKM pola suma de FPKMs que hai nunha mostra.[77][78][79]
- Rendemento de ARN total na mostra: como se extrae a mesma cantidade de ARN de cada mostra, as mostras con máis ARN total terán menos ARN por xene. Estes xenes parecen ter unha expresión diminuída, orixinando falsos positivos nas análises posteriores "augas abaixo".[75]
- Varianza para a expresión de cada xene: esta é modelada para ter en conta o erro de mostraxe (importante para xenes con baixos recontos de lecturas), aumentar a potencia e diminuír os falsos positivos. A varianza pode estimarse como unha distribución normal, de Poisson ou binomial negativa[80][81][82] e é frecuentemente descomposta en varianza técnica e varianza biolóxica.
Cuantificación absoluta
[editar | editar a fonte]A cuantificación absoluta da expresión xénica non é posible coa maioría dos experimentos de RNA-Seq, que cuantifican a expresión relativa a todos os transcritos. É posible realizar unha RNA-Seq con spike-ins en mostras de ARN a concentracións coñecidas. Despois de secuenciar, os recontos de lecturas de secuencias spike-in utilízanse para determinar as relacións entre os recontos das lecturas de cada xene e as cantidades absolutas de fragmentos biolóxicos.[11][83] Nun exemplo, esta técnica foi utilizada en embrións do anfibio Xenopus tropicalis para determinar a cinética de transcrición.[84]
Expresión diferencial
[editar | editar a fonte]O uso máis simple pero xeralmente o máis potente da RNA-Seq é atopar diferenzas na expresión xénica entre dúas ou máis condicións (por exemplo, suxeito tratado ou non tratado); este proceso denomínase expresión diferencial. Os resultados refírense frecuentemente a xenes expresados diferencialmente (DEGs) e estes xenes poden estar regulados á alza ou á baixa (é dicir, máis altos ou máis baixos na condición de interese). Hai moitas ferramentas que realizan esta expresión diferencial. A maioría funcionan en linguaxe de programación R, Python ou a liña de comando Unix. Entre as ferramentas usadas comunmente están DESeq,[81] edgeR,[82] e voom+limma,[80][85] todas as cales están dispoñibles por medio de R/Bioconductor.[86][87] Estas son as consideracións comúns a ter en conta cando se realiza a expresión diferencial:
- Entradas (inputs): as entradas en expresión diferencial inclúen: (1) unha matriz de expresión de RNA-Seq (M xenes × N mostras) e (2) unha matriz de deseño que contén as condicións experimentais para N mostras. A matriz de deseño máis simple contén unha columna, que corresponde a etiquetas para a condición que se está a comprobar. Outros covariados (tamén denominados factores, características, etiquetas ou parámetros) poden incluír efectos en lote, coñecidos como artefactos e calquera metadato que puidese confundir ou mediar a expresión xénica. Ademais dos covariados coñecidos, os covariados descoñecidos poden tamén estimarse por estratexias de aprendizaxe de máquina non supervisada como análises de compoñente principal, variable surrogada,[88] e PEER.[47] A miúdo empréganse análises de variable oculta para datos de RNA-Seq de tecidos humanos, que tipicamente teñen artefactos adicionais non capturados nos metadatos (por exemplo, o tempo isquémico, fontes de datos de múltiples institucións, características clínicas subxacentes, recollida de datos ao longo de moitos anos con moito persoal implicado).
- Métodos: a maioría das ferramentas usan a regresión ou estatística non paramétrica para identificar xenes expresados diferencialmente e están ou baseados en reconto (DESeq2, limma, edgeR) ou baseados en ensamblaxe (por cuantificación sen aliñamento, sleuth (indagación),[89] Cuffdiff,[90] Ballgown[91]).[92] Seguindo a regresión, a maioría das ferramentas empregan axustes de valor p de taxa de erro por familia (FWER) ou taxa de descubrimento falso (FDR) para considerar hipóteses múltiples (en estudos humanos, ~20000 xenes codificantes de proteínas ou ~50000 biotipos).
- Resultados (outputs): un resultdo típico consiste en filas que corresponden co número de xenes e polo menos tres columnas, o logaritmo do "cambio de veces" (log fold change) de cada xene (transformada logarítmica da proporción de expresión entre condicións, unha medida do tamaño do efecto), valor p, e valor p axustado para comparacións múltiples. Os xenes son definidos como bioloxicamente significativos se pasan os puntos de corte para o tamaño do efecto (logaritmo do "cambio de veces") e significación estatística. Idealmente, estes puntos de corte deberían ser especificados a priori, pero a natureza dos experimentos de RNA-Seq é con frecuencia exploratoria polo que é difícil predicir os tamaños do efecto e os pertinentes cortes con antelación.
- Inconvenientes: a razón de ser destes métodos complexos é evitar os milleiros de dificultades que poden levar a cometer erros estatísticos e interpretacións enganosas. Entre estas dificultades están o incremento das taxas de falsos positivos (debido a comparacións múltiples), artefactos da preparación das mostras, heteroxeneidade da mostra (como fondos xenéticos mesturados), mostras altamente correlacionadas, non ter en conta deseños experimentais multinivel e un mal deseño experimental. Un notable inconveniente é ver os resultados en Microsoft Excel.[93] Aínda que é un programa cómodo, Excel converte automaticamente algúns nomes de xenes (SEPT1, DEC1, MARCH2) en datos ou números con comas flotantes.
- Elección de ferramentas e punto de referencia: hai numerosos xeitos de comparar os resultados destas ferramentas, e DESeq2 adoita ser algo mellor que outros métodos.[92][94][95][96][97][98][99] Igual que con outros métodos, a elección do punto de referencia (benchmarking) consiste en comparar os resultados das ferramentas unhas con outras e con estándares de referencia coñecidos.
As análises posteriores (augas abaixo) dunha lista de xenes expresados diferencialmente son de dous tipos: validar as observacións e facer inferencias biolóxicas. Debido aos inconvenientes da expresión diferencial e a RNA-Seq, as observacións importantes son replicadas con: (1) un método ortogonal nas mesmas mostras (como a PCR en tempo real) ou (2) outro experimento, ás veces prerrexistrado, nunha nova cohorte. Este último axuda a asegurarse da xeneralizabilidade e pode normalmente ir seguido dunha metaanálise de todas as cohortes do pool. O método máis común para obter unha comprensión biolóxica de nivel máis alto dos resultados é a análise de enriquecemento do conxunto de xenes, aínda que ás veces se empregan estratexias de xenes candidatos. O enriquecemento do conxunto de xenes determina se o solapamento entre dous conxuntos de xenes é estatisticamente significativo; nese caso vese o solapamento entre xenes expresados diferencialmente e conxuntos de xenes a partir de vías/bases de datos coñecidas (por exemplo, Gene Ontology, KEGG, Human Phenotype Ontology) ou a partir de análises complementarias dos mesmos datos (como as redes de coexpresión). Ferramentas comúns para o enriquecemento de conxuntos de xenes son interfaces web (por exemplo, ENRICHR, g:profiler) e paquetes de software. Cando se avalían os resultados de enriquecemento, unha heurística uilizada é primeiro buscar o enriquecemento da bioloxía coñecida como comprobación sensata e despois expandir o alcance para buscar bioloxía nova.

Empalme alternativo
[editar | editar a fonte]O empalme de ARN é un proceso típico dos eucariotas e contribúe significativamente á regulación de proteínas e a súa diversidade, e ocorre en >90% dos xenes humanos.[100] Hai múltiples modos de empalme alternativo: omisión de exón (o modo de empalme máis común en humanos e eucariotas superiores), exóns mutuamente excluíntes, doante alternativo ou sitios aceptores, retención de exóns (o modo de empalme máis común en plantas, fungos e protozoos), sitio alternativo de comezo da transcrición (promotor), e poliadenilación alternativa.[100] Un obxectivo da RNA-Seq é identificar eventos de empalme alternativo e comprobar se difiren entre distintas condicións. A secuenciación de lecturas longas captura o transcrito completo e así minimiza moitos dos problemas que se presentan na estimación da abundancia de isoformas, como o mapado de lecturas ambiguas. Para a RNA-Seq de lecturas curtas, hai múltiples métodos para detectar o empalme alternativo que pode ser clasificado en tres grupos principais:[101][102][103]
- Baseado no reconto (tamén baseado no evento, empalme diferencial): estima a retención de exóns. Exemplos son DEXSeq,[104] MATS,[105] e SeqGSEA.[106]
- Baseado en isoformas (tamén módulos de multilectura, expresión de isoformas diferencial): estima primeiro a abundancia de isoformas e despois a abundancia relativa entre condicións. Exemplos son Cufflinks 2[107] e DiffSplice.[108]
- Baseado na escisión de intróns: calcula o empalme alternativo usando lecturas que foron divididas. Exemplos son MAJIQ[109] e Leafcutter.[103]
As ferramentas de expresión diferencial de xenes poden utilizarse tamén para a expresión diferencial de isoformas se as isoformas están cuantificadas con antelación con outras ferramentas como RSEM.[110]
Redes de coexpresión
[editar | editar a fonte]As redes de coexpresión son representacións derivadas de datos de xenes que se comportan de maneira similar en distintos tecidos e distintas condicións experimentais.[111] O seu principal propósito é a xeración de hipóteses e estratexias de "culpa por asociación" para inferir as funcións de xenes previamente descoñecidos.[111] Os datos de RNA-Seq foron utiizados para inferir xenes implicados en vías específicas baseándose na correlación de Pearson, tanto en plantas[112] coma en mamíferos.[113] A principal vantaxe dos datos de RNA-Seq neste tipo de análises sobre as plataformas de micromatrices é a capacidade de cubrir o transcritoma enteiro, permitindo, por tanto, a posibilidade de obter representacións máis completas das redes regulatorias de xenes. A regulación diferencial das isoformas de empalme do mesmo xene pode detectarse e usarse para predicir e saber as súas funcións biolóxicas.[114][115] Utilizáronse con éxito a análises de redes de coexpresión de xenes ponderada para identificar módulos de coexpresión e xenes centrais intramodulares baseándose en datos de RNA seq. Os módulos de coexpresión poden corresponder a tipos celulares ou vías. Os centros intramodulares altamente conectados poden ser interpretados como representativos dos seus respectivos módulos. Un eigengene é unha suma ponderada da expresión de todos os xenes nun módulo. Os eigengenes son biomarcadores útiles (features) para a diagnose e prognóstico.[116] Propuxéronse estratexias de Transformación Estabilizadora da Varianza para estimar os coeficientes de correlación baseados en datos de RNA seq.[112]
Descubrimento de variantes
[editar | editar a fonte]A RNA-Seq captura a variación no ADN, incluíndo as variantes de nucleótido único, pequenas insercións/delecións, e as variacións estruturais. A Variant calling en RNA-Seq é similar ao callind de variantes de ADN e a miúdo emprega as mesmas ferramentas (como SAMtools mpileup[117] e GATK HaplotypeCaller[118]) con axustes para te en conta o empalme. Unha dimensión única das variantes de ARN é expresión específica de alelo (ASE): asvariantes dun só haplotipo poderían expresarse preferencialmente debido a efectos reguladores como a impresión xenómica e a expresión de loci de trazos cuantitativos e as variantes raras non codificantes.[119][120] Entre as limitacións na identificación de variantes de ARN está que só reflicte rexións expresadas (en humanos <5% do xenoma) e ten unha calidade peor cando a comparamos coa secuenciación de ADN directa.
Edición do ARN (alteracións post-transcricionais)
[editar | editar a fonte]- Véxase tamén: Edición do ARN.
Ter as secuencias transcritómicas e xenómicas que se corresponden entre si dun individuo pode axudar a detectar edicións post-transcricionais (edición do ARN).[3] Un evento de modificación post-transcricional é identificado se o transcito do xene ten un alelo/variante non observada nos datos xenómicos.

Detección de xenes de fusión
[editar | editar a fonte]- Véxase tamén: Xene de fusión.
Os xenes de fusión, causados por diferentes modificacions estruturais no xenoma, están recibindo moita atención debido á súa relación co cancro.[121] A capacidade da RNA-Seq de analizar o transcritoma completo dunha mostra de modo non nesgado faina unha ferramenta atractiva para buscar este tipo de eventos comúns no cancro.[4]
A idea dedúcese do proceso de aliñar as lecturas transcritómicas curtas cun xenma de referencia. A maioría das lecturas curtas están comprendidas dentro dun exón completo, e esperaríase que un conxunto menor pero aínda grande se mapase a unións exón-exón coñecidas. O resto das lecturas curtas non mapadas serían despois analizadas para determinar se coinciden con unións exón-exón nas que os exóns proceden de diferentes xenes. Isto sería unha evidencia dun posible evento de fusión, pero, debido á lonxitude das lecturas, isto podería resultar moi ruidoso. Unha aproximación alternativa é usar lecturas de extremos apareados, cun número potencialmente grande de lecturas apareadas que se maparían a un exón diferente, dando unha mellor cobertura para estes eventos (ver figura). Non obstante, o resultado final consiste en combinacións múltiples e potencialmente novas de xenes que proporcionan un punto de comezo ideal para ulteriores validacións.
Historia
[editar | editar a fonte].png?lang=gl)
A RNA-Seq empezou a desenvolverse a metade da década de 2000 coa aparición da tecnoloxía de secuenciación de seguinte xeración.[122] Os primeiros artigos que falaban da RNA-Seq aínda que sen usar aínda o termo trataban sobre liñas celulares do cancro de próstata[123] (datadas en 2006), a planta Medicago truncatula[124] (2006), o millo[125] (2007), e Arabidopsis thaliana[126] (2007), mentres que o termo "RNA-Seq" foi mencionado por primeira vez en 2008.[127] O número de artigos que se refiren á RNA-Seq no título ou no resumo (na figura a liña azul) está incrementándose constantemente e en 2018 publicáranse 6754 artigos (ligazón á busca de PubMed).
Aplicacións á medicina
[editar | editar a fonte]A RNA-Seq ten o potential de identificar nova bioloxía sobre doenzas, perfilar biomarcadores para indicacións clínicas, inferir vías tratables con fármacos e facer diagnósticos xenéticos. Estes resultados poderían despois ser personalizados para subgrupos de pacientes ou incluso individuos, o que serviría para facer unha prevención máis efectiva e mellores diagnósticos e terapias. A aplicabilidade destas estratexias está en parte determinada polo custo en diñeiro e tempo; unha limitación importante é o tempo que cómpre para que o equipo de especialistas (bioinformáticos, médicos, investigadores básicos e técnicos) interpreten completamente a enorme cantidade de datos xerados por estas análises.
Iniciativas de secuenciación a grande escala
[editar | editar a fonte]Estáselle dando grande importancia aos datos de RNA-Seq desde que os proxectos Encyclopedia of DNA Elements (ENCODE) e The Cancer Genome Atlas (TCGA) utilizaron este enfoque para caracterizar ducias de liñas celulares[128] e miles de mostras de tumores primarias,[129] respectivamente. ENCODE trata de identificar rexións regulatorias en todo o xenoma en diferentes cohortes de liñas celulares e os datos transcritómicos son fundamentais para entender os efectos augas abaixo destas capas regulatorias xenética e epixenética. Ao contraio, o TCGA trata de recoller e analizar milleiros de mostras tomadas de pacientes de 30 tipos de tumores para comprender os mecanismos subxacentes da transformación maligna e a súa progresión. Neste contexto os datos de RNA-Seq proporcionan unha instantánea única do status transcritómico da doenza e examinan unha poboación non nesgada de transcritos que posibilita a identificación de novos transcritos, transcritos de fusión e ARN non codificante, que poderían ser indetectados con outras tecnoloxías.
Notas
[editar | editar a fonte]- ↑ Shafee T, Lowe R (2017). "Eukaryotic and prokaryotic gene structure". WikiJournal of Medicine (en inglés) 4 (1). doi:10.15347/wjm/2017.002.
- ↑ Chu Y, Corey DR (August 2012). "RNA sequencing: platform selection, experimental design, and data interpretation". Nucleic Acid Therapeutics 22 (4): 271–4. PMC 3426205. PMID 22830413. doi:10.1089/nat.2012.0367.
- ↑ 3,0 3,1 3,2 Wang Z, Gerstein M, Snyder M (January 2009). "RNA-Seq: a revolutionary tool for transcriptomics". Nature Reviews. Genetics 10 (1): 57–63. PMC 2949280. PMID 19015660. doi:10.1038/nrg2484.
- ↑ 4,0 4,1 Maher CA, Kumar-Sinha C, Cao X, Kalyana-Sundaram S, Han B, Jing X, et al. (March 2009). "Transcriptome sequencing to detect gene fusions in cancer". Nature 458 (7234): 97–101. Bibcode:2009Natur.458...97M. PMC 2725402. PMID 19136943. doi:10.1038/nature07638.
- ↑ Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS (July 2012). "The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments". Nature Protocols 7 (8): 1534–50. PMC 3535016. PMID 22836135. doi:10.1038/nprot.2012.086.
- ↑ Lee JH, Daugharthy ER, Scheiman J, Kalhor R, Yang JL, Ferrante TC, et al. (March 2014). "Highly multiplexed subcellular RNA sequencing in situ". Science 343 (6177): 1360–3. Bibcode:2014Sci...343.1360L. PMC 4140943. PMID 24578530. doi:10.1126/science.1250212.
- ↑ Kukurba KR, Montgomery SB (April 2015). "RNA Sequencing and Analysis". Cold Spring Harbor Protocols 2015 (11): 951–69. PMC 4863231. PMID 25870306. doi:10.1101/pdb.top084970.
- ↑ 8,0 8,1 8,2 8,3 8,4 Griffith M, Walker JR, Spies NC, Ainscough BJ, Griffith OL (August 2015). "Informatics for RNA Sequencing: A Web Resource for Analysis on the Cloud". PLoS Computational Biology 11 (8): e1004393. Bibcode:2015PLSCB..11E4393G. PMC 4527835. PMID 26248053. doi:10.1371/journal.pcbi.1004393.
- ↑ "RNA-seqlopedia". rnaseq.uoregon.edu. Consultado o 2017-02-08.
- ↑ Morin R, Bainbridge M, Fejes A, Hirst M, Krzywinski M, Pugh T, et al. (July 2008). "Profiling the HeLa S3 transcriptome using randomly primed cDNA and massively parallel short-read sequencing". BioTechniques 45 (1): 81–94. PMID 18611170. doi:10.2144/000112900. Arquivado dende o orixinal o 21 de marzo de 2009. Consultado o 30 de decembro de 2019.
- ↑ 11,0 11,1 11,2 Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (July 2008). "Mapping and quantifying mammalian transcriptomes by RNA-Seq". Nature Methods 5 (7): 621–8. PMID 18516045. doi:10.1038/nmeth.1226.
- ↑ Chen EA, Souaiaia T, Herstein JS, Evgrafov OV, Spitsyna VN, Rebolini DF, Knowles JA (October 2014). "Effect of RNA integrity on uniquely mapped reads in RNA-Seq". BMC Research Notes 7 (1): 753. PMC 4213542. PMID 25339126. doi:10.1186/1756-0500-7-753.
- ↑ Liu D, Graber JH (February 2006). "Quantitative comparison of EST libraries requires compensation for systematic biases in cDNA generation". BMC Bioinformatics 7: 77. PMC 1431573. PMID 16503995. doi:10.1186/1471-2105-7-77.
- ↑ Garalde DR, Snell EA, Jachimowicz D, Sipos B, Lloyd JH, Bruce M, et al. (March 2018). "Highly parallel direct RNA sequencing on an array of nanopores". Nature Methods 15 (3): 201–206. PMID 29334379. doi:10.1038/nmeth.4577.
- ↑ 15,0 15,1 "Shapiro E, Biezuner T, Linnarsson S (September 2013). "Single-cell sequencing-based technologies will revolutionize whole-organism science". Nature Reviews. Genetics 14 (9): 618–30. PMID 23897237. doi:10.1038/nrg3542."
- ↑ Kolodziejczyk AA, Kim JK, Svensson V, Marioni JC, Teichmann SA (May 2015). "The technology and biology of single-cell RNA sequencing". Molecular Cell 58 (4): 610–20. PMID 26000846. doi:10.1016/j.molcel.2015.04.005.
- ↑ Montoro DT, Haber AL, Biton M, Vinarsky V, Lin B, Birket SE, et al. (August 2018). "A revised airway epithelial hierarchy includes CFTR-expressing ionocytes". Nature 560 (7718): 319–324. Bibcode:2018Natur.560..319M. PMC 6295155. PMID 30069044. doi:10.1038/s41586-018-0393-7.
- ↑ Plasschaert LW, Žilionis R, Choo-Wing R, Savova V, Knehr J, Roma G, et al. (August 2018). "A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte". Nature 560 (7718): 377–381. Bibcode:2018Natur.560..377P. PMC 6108322. PMID 30069046. doi:10.1038/s41586-018-0394-6.
- ↑ Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, et al. (May 2015). "Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells". Cell 161 (5): 1187–1201. PMC 4441768. PMID 26000487. doi:10.1016/j.cell.2015.04.044.
- ↑ Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, et al. (May 2015). "Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets". Cell 161 (5): 1202–1214. PMC 4481139. PMID 26000488. doi:10.1016/j.cell.2015.05.002.
- ↑ "Hebenstreit D (November 2012). "Methods, Challenges and Potentials of Single Cell RNA-seq". Biology 1 (3): 658–67. PMC 4009822. PMID 24832513. doi:10.3390/biology1030658."
- ↑ Eberwine J, Sul JY, Bartfai T, Kim J (January 2014). "The promise of single-cell sequencing". Nature Methods 11 (1): 25–7. PMID 24524134. doi:10.1038/nmeth.2769.
- ↑ Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, et al. (May 2009). "mRNA-Seq whole-transcriptome analysis of a single cell". Nature Methods 6 (5): 377–82. PMID 19349980. doi:10.1038/NMETH.1315.
- ↑ Islam S, Kjällquist U, Moliner A, Zajac P, Fan JB, Lönnerberg P, Linnarsson S (July 2011). "Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq". Genome Research 21 (7): 1160–7. PMC 3129258. PMID 21543516. doi:10.1101/gr.110882.110.
- ↑ Ramsköld D, Luo S, Wang YC, Li R, Deng Q, Faridani OR, et al. (August 2012). "Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells". Nature Biotechnology 30 (8): 777–82. PMC 3467340. PMID 22820318. doi:10.1038/nbt.2282.
- ↑ Hashimshony T, Wagner F, Sher N, Yanai I (September 2012). "CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification". Cell Reports 2 (3): 666–73. PMID 22939981. doi:10.1016/j.celrep.2012.08.003.
- ↑ Singh M, Al-Eryani G, Carswell S, Ferguson JM, Blackburn J, Barton K, Roden D, Luciani F, Phan T, Junankar S, Jackson K, Goodnow CC, Smith MA, Swarbrick A (2018). "High-throughput targeted long-read single cell sequencing reveals the clonal and transcriptional landscape of lymphocytes". bioRxiv. doi:10.1101/424945.
- ↑ Sasagawa Y, Nikaido I, Hayashi T, Danno H, Uno KD, Imai T, Ueda HR (April 2013). "Quartz-Seq: a highly reproducible and sensitive single-cell RNA sequencing method, reveals non-genetic gene-expression heterogeneity". Genome Biology 14 (4): R31. PMC 4054835. PMID 23594475. doi:10.1186/gb-2013-14-4-r31.
- ↑ Kouno T, Moody J, Kwon AT, Shibayama Y, Kato S, Huang Y, et al. (January 2019). "C1 CAGE detects transcription start sites and enhancer activity at single-cell resolution". Nature Communications 10 (1): 360. Bibcode:2019NatCo..10..360K. PMC 6341120. PMID 30664627. doi:10.1038/s41467-018-08126-5.
- ↑ Dal Molin A, Di Camillo B (January 2018). "How to design a single-cell RNA-sequencing experiment: pitfalls, challenges and perspectives". Briefings in Bioinformatics: bby007. PMID 29394315. doi:10.1093/bib/bby007.
- ↑ Peterson VM, Zhang KX, Kumar N, Wong J, Li L, Wilson DC, et al. (October 2017). "Multiplexed quantification of proteins and transcripts in single cells". Nature Biotechnology 35 (10): 936–939. PMID 28854175. doi:10.1038/nbt.3973.
- ↑ Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, et al. (September 2017). "Simultaneous epitope and transcriptome measurement in single cells". Nature Methods 14 (9): 865–868. PMC 5669064. PMID 28759029. doi:10.1038/nmeth.4380.
- ↑ Raj B, Wagner DE, McKenna A, Pandey S, Klein AM, Shendure J, et al. (June 2018). "Simultaneous single-cell profiling of lineages and cell types in the vertebrate brain". Nature Biotechnology 36 (5): 442–450. PMC 5938111. PMID 29608178. doi:10.1038/nbt.4103.
- ↑ Olmos D, Arkenau HT, Ang JE, Ledaki I, Attard G, Carden CP, et al. (January 2009). "Circulating tumour cell (CTC) counts as intermediate end points in castration-resistant prostate cancer (CRPC): a single-centre experience". Annals of Oncology 20 (1): 27–33. PMID 18695026. doi:10.1093/annonc/mdn544.
- ↑ Levitin HM, Yuan J, Sims PA (April 2018). "Single-Cell Transcriptomic Analysis of Tumor Heterogeneity". Trends in Cancer (en English) 4 (4): 264–268. PMC 5993208. PMID 29606308. doi:10.1016/j.trecan.2018.02.003.
- ↑ Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, et al. (November 2018). "A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade". Cell (en English) 175 (4): 984–997.e24. PMC 6410377. PMID 30388455. doi:10.1016/j.cell.2018.09.006.
- ↑ Stephenson W, Donlin LT, Butler A, Rozo C, Bracken B, Rashidfarrokhi A, et al. (February 2018). "Single-cell RNA-seq of rheumatoid arthritis synovial tissue using low-cost microfluidic instrumentation". Nature Communications 9 (1): 791. Bibcode:2018NatCo...9..791S. PMC 5824814. PMID 29476078. doi:10.1038/s41467-017-02659-x.
- ↑ Avraham R, Haseley N, Brown D, Penaranda C, Jijon HB, Trombetta JJ, et al. (September 2015). "Pathogen Cell-to-Cell Variability Drives Heterogeneity in Host Immune Responses". Cell 162 (6): 1309–21. PMC 4578813. PMID 26343579. doi:10.1016/j.cell.2015.08.027.
- ↑ Cao J, Packer JS, Ramani V, Cusanovich DA, Huynh C, Daza R, et al. (August 2017). "Comprehensive single-cell transcriptional profiling of a multicellular organism". Science 357 (6352): 661–667. Bibcode:2017Sci...357..661C. PMC 5894354. PMID 28818938. doi:10.1126/science.aam8940.
- ↑ Plass M, Solana J, Wolf FA, Ayoub S, Misios A, Glažar P, et al. (May 2018). "Cell type atlas and lineage tree of a whole complex animal by single-cell transcriptomics". Science 360 (6391): eaaq1723. PMID 29674432. doi:10.1126/science.aaq1723. Arquivado dende o orixinal o 11 de maio de 2020. Consultado o 30 de decembro de 2019.
- ↑ Fincher CT, Wurtzel O, de Hoog T, Kravarik KM, Reddien PW (May 2018). "Schmidtea mediterranea". Science 360 (6391): eaaq1736. PMC 6563842. PMID 29674431. doi:10.1126/science.aaq1736.
- ↑ Wagner DE, Weinreb C, Collins ZM, Briggs JA, Megason SG, Klein AM (June 2018). "Single-cell mapping of gene expression landscapes and lineage in the zebrafish embryo". Science 360 (6392): 981–987. Bibcode:2018Sci...360..981W. PMC 6083445. PMID 29700229. doi:10.1126/science.aar4362.
- ↑ Farrell JA, Wang Y, Riesenfeld SJ, Shekhar K, Regev A, Schier AF (June 2018). "Single-cell reconstruction of developmental trajectories during zebrafish embryogenesis". Science 360 (6392): eaar3131. PMC 6247916. PMID 29700225. doi:10.1126/science.aar3131.
- ↑ Briggs JA, Weinreb C, Wagner DE, Megason S, Peshkin L, Kirschner MW, Klein AM (June 2018). "The dynamics of gene expression in vertebrate embryogenesis at single-cell resolution". Science 360 (6392): eaar5780. PMC 6038144. PMID 29700227. doi:10.1126/science.aar5780.
- ↑ You J. "Science's 2018 Breakthrough of the Year: tracking development cell by cell". Science Magazine. American Association for the Advancement of Science.
- ↑ 46,0 46,1 Li H, Lovci MT, Kwon YS, Rosenfeld MG, Fu XD, Yeo GW (December 2008). "Determination of tag density required for digital transcriptome analysis: application to an androgen-sensitive prostate cancer model". Proceedings of the National Academy of Sciences of the United States of America 105 (51): 20179–84. Bibcode:2008PNAS..10520179L. PMC 2603435. PMID 19088194. doi:10.1073/pnas.0807121105.
- ↑ 47,0 47,1 Stegle O, Parts L, Piipari M, Winn J, Durbin R (February 2012). "Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses". Nature Protocols 7 (3): 500–7. PMC 3398141. PMID 22343431. doi:10.1038/nprot.2011.457.
- ↑ Kingsford C, Patro R (June 2015). "Reference-based compression of short-read sequences using path encoding". Bioinformatics 31 (12): 1920–8. PMC 4481695. PMID 25649622. doi:10.1093/bioinformatics/btv071.
- ↑ 49,0 49,1 Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. (May 2011). "Full-length transcriptome assembly from RNA-Seq data without a reference genome". Nature Biotechnology 29 (7): 644–52. PMC 3571712. PMID 21572440. doi:10.1038/nbt.1883.
- ↑ "De Novo Assembly Using Illumina Reads" (PDF). Consultado o 22 October 2016.
- ↑ Zerbino DR, Birney E (May 2008). "Velvet: algorithms for de novo short read assembly using de Bruijn graphs". Genome Research 18 (5): 821–9. PMC 2336801. PMID 18349386. doi:10.1101/gr.074492.107.
- ↑ "Oases: a transcriptome assembler for very short reads". Arquivado dende o orixinal o 29 de novembro de 2018. Consultado o 30 de decembro de 2019.
- ↑ Chang Z, Li G, Liu J, Zhang Y, Ashby C, Liu D, et al. (February 2015). "Bridger: a new framework for de novo transcriptome assembly using RNA-seq data". Genome Biology 16 (1): 30. PMC 4342890. PMID 25723335. doi:10.1186/s13059-015-0596-2.
- ↑ 54,0 54,1 Li B, Fillmore N, Bai Y, Collins M, Thomson JA, Stewart R, Dewey CN (December 2014). "Evaluation of de novo transcriptome assemblies from RNA-Seq data". Genome Biology 15 (12): 553. PMC 4298084. PMID 25608678. doi:10.1186/s13059-014-0553-5.
- ↑ 55,0 55,1 Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. (January 2013). "STAR: ultrafast universal RNA-seq aligner". Bioinformatics 29 (1): 15–21. PMC 3530905. PMID 23104886. doi:10.1093/bioinformatics/bts635.
- ↑ Langmead B, Trapnell C, Pop M, Salzberg SL (2009). "Ultrafast and memory-efficient alignment of short DNA sequences to the human genome". Genome Biology 10 (3): R25. PMC 2690996. PMID 19261174. doi:10.1186/gb-2009-10-3-r25.
- ↑ Trapnell C, Pachter L, Salzberg SL (May 2009). "TopHat: discovering splice junctions with RNA-Seq". Bioinformatics 25 (9): 1105–11. PMC 2672628. PMID 19289445. doi:10.1093/bioinformatics/btp120.
- ↑ Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. (March 2012). "Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks". Nature Protocols 7 (3): 562–78. PMC 3334321. PMID 22383036. doi:10.1038/nprot.2012.016.
- ↑ Liao Y, Smyth GK, Shi W (May 2013). "The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote". Nucleic Acids Research 41 (10): e108. PMC 3664803. PMID 23558742. doi:10.1093/nar/gkt214.
- ↑ Kim D, Langmead B, Salzberg SL (April 2015). "HISAT: a fast spliced aligner with low memory requirements". Nature Methods 12 (4): 357–60. PMC 4655817. PMID 25751142. doi:10.1038/nmeth.3317.
- ↑ Patro R, Mount SM, Kingsford C (May 2014). "Sailfish enables alignment-free isoform quantification from RNA-seq reads using lightweight algorithms". Nature Biotechnology 32 (5): 462–4. PMC 4077321. PMID 24752080. arXiv:1308.3700. doi:10.1038/nbt.2862.
- ↑ Bray NL, Pimentel H, Melsted P, Pachter L (May 2016). "Near-optimal probabilistic RNA-seq quantification". Nature Biotechnology 34 (5): 525–7. PMID 27043002. doi:10.1038/nbt.3519.
- ↑ Wu TD, Watanabe CK (May 2005). "GMAP: a genomic mapping and alignment program for mRNA and EST sequences". Bioinformatics 21 (9): 1859–75. PMID 15728110. doi:10.1093/bioinformatics/bti310.
- ↑ Baruzzo G, Hayer KE, Kim EJ, Di Camillo B, FitzGerald GA, Grant GR (February 2017). "Simulation-based comprehensive benchmarking of RNA-seq aligners". Nature Methods (en inglés) 14 (2): 135–139. PMC 5792058. PMID 27941783. doi:10.1038/nmeth.4106.
- ↑ Engström PG, Steijger T, Sipos B, Grant GR, Kahles A, Rätsch G, et al. (December 2013). "Systematic evaluation of spliced alignment programs for RNA-seq data". Nature Methods (en inglés) 10 (12): 1185–91. PMC 4018468. PMID 24185836. doi:10.1038/nmeth.2722.
- ↑ Lu B, Zeng Z, Shi T (February 2013). "Comparative study of de novo assembly and genome-guided assembly strategies for transcriptome reconstruction based on RNA-Seq". Science China Life Sciences 56 (2): 143–55. PMID 23393030. doi:10.1007/s11427-013-4442-z.
- ↑ Bradnam KR, Fass JN, Alexandrov A, Baranay P, Bechner M, Birol I, et al. (July 2013). "Assemblathon 2: evaluating de novo methods of genome assembly in three vertebrate species". GigaScience 2 (1): 10. Bibcode:2013arXiv1301.5406B. PMC 3844414. PMID 23870653. arXiv:1301.5406. doi:10.1186/2047-217X-2-10.
- ↑ Greenbaum D, Colangelo C, Williams K, Gerstein M (2003). "Comparing protein abundance and mRNA expression levels on a genomic scale". Genome Biology 4 (9): 117. PMC 193646. PMID 12952525. doi:10.1186/gb-2003-4-9-117.
- ↑ Zhang ZH, Jhaveri DJ, Marshall VM, Bauer DC, Edson J, Narayanan RK, et al. (August 2014). "A comparative study of techniques for differential expression analysis on RNA-Seq data". PLOS ONE 9 (8): e103207. Bibcode:2014PLoSO...9j3207Z. PMC 4132098. PMID 25119138. doi:10.1371/journal.pone.0103207.
- ↑ Anders S, Pyl PT, Huber W (January 2015). "HTSeq--a Python framework to work with high-throughput sequencing data". Bioinformatics 31 (2): 166–9. PMC 4287950. PMID 25260700. doi:10.1093/bioinformatics/btu638.
- ↑ Liao Y, Smyth GK, Shi W (April 2014). "featureCounts: an efficient general purpose program for assigning sequence reads to genomic features". Bioinformatics 30 (7): 923–30. PMID 24227677. arXiv:1305.3347. doi:10.1093/bioinformatics/btt656.
- ↑ Schmid MW, Grossniklaus U (February 2015). "Rcount: simple and flexible RNA-Seq read counting". Bioinformatics 31 (3): 436–7. PMID 25322836. doi:10.1093/bioinformatics/btu680.
- ↑ Finotello F, Lavezzo E, Bianco L, Barzon L, Mazzon P, Fontana P, et al. (2014). "Reducing bias in RNA sequencing data: a novel approach to compute counts". BMC Bioinformatics. 15 Suppl 1 (Suppl 1): S7. PMC 4016203. PMID 24564404. doi:10.1186/1471-2105-15-s1-s7.
- ↑ Hashimoto TB, Edwards MD, Gifford DK (March 2014). "Universal count correction for high-throughput sequencing". PLoS Computational Biology 10 (3): e1003494. Bibcode:2014PLSCB..10E3494H. PMC 3945112. PMID 24603409. doi:10.1371/journal.pcbi.1003494.
- ↑ 75,0 75,1 Robinson MD, Oshlack A (2010). "A scaling normalization method for differential expression analysis of RNA-seq data". Genome Biology 11 (3): R25. PMC 2864565. PMID 20196867. doi:10.1186/gb-2010-11-3-r25.
- ↑ Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. (May 2010). "Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation". Nature Biotechnology 28 (5): 511–5. PMC 3146043. PMID 20436464. doi:10.1038/nbt.1621.
- ↑ Pachter L (19 April 2011). "Models for transcript quantification from RNA-Seq". arXiv:1104.3889 q-bio.GN.
- ↑ "What the FPKM? A review of RNA-Seq expression units". The farrago. 8 May 2014. Consultado o 28 March 2018.
- ↑ Wagner GP, Kin K, Lynch VJ (December 2012). "Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples". Theory in Biosciences = Theorie in den Biowissenschaften 131 (4): 281–5. PMID 22872506. doi:10.1007/s12064-012-0162-3.
- ↑ 80,0 80,1 Law CW, Chen Y, Shi W, Smyth GK (February 2014). "voom: Precision weights unlock linear model analysis tools for RNA-seq read counts". Genome Biology 15 (2): R29. PMC 4053721. PMID 24485249. doi:10.1186/gb-2014-15-2-r29.
- ↑ 81,0 81,1 Anders S, Huber W (2010). "Differential expression analysis for sequence count data". Genome Biology 11 (10): R106. PMC 3218662. PMID 20979621. doi:10.1186/gb-2010-11-10-r106.
- ↑ 82,0 82,1 Robinson MD, McCarthy DJ, Smyth GK (January 2010). "edgeR: a Bioconductor package for differential expression analysis of digital gene expression data". Bioinformatics 26 (1): 139–40. PMC 2796818. PMID 19910308. doi:10.1093/bioinformatics/btp616.
- ↑ Marguerat S, Schmidt A, Codlin S, Chen W, Aebersold R, Bähler J (October 2012). "Quantitative analysis of fission yeast transcriptomes and proteomes in proliferating and quiescent cells". Cell 151 (3): 671–83. PMC 3482660. PMID 23101633. doi:10.1016/j.cell.2012.09.019.
- ↑ Owens ND, Blitz IL, Lane MA, Patrushev I, Overton JD, Gilchrist MJ, et al. (January 2016). "Measuring Absolute RNA Copy Numbers at High Temporal Resolution Reveals Transcriptome Kinetics in Development". Cell Reports 14 (3): 632–647. PMC 4731879. PMID 26774488. doi:10.1016/j.celrep.2015.12.050.
- ↑ Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (April 2015). "limma powers differential expression analyses for RNA-sequencing and microarray studies". Nucleic Acids Research 43 (7): e47. PMC 4402510. PMID 25605792. doi:10.1093/nar/gkv007.
- ↑ "Bioconductor - Open source software for bioinformatics".
- ↑ Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, et al. (February 2015). "Orchestrating high-throughput genomic analysis with Bioconductor". Nature Methods 12 (2): 115–21. PMC 4509590. PMID 25633503. doi:10.1038/nmeth.3252.
- ↑ Leek JT, Storey JD (September 2007). "Capturing heterogeneity in gene expression studies by surrogate variable analysis". PLoS Genetics 3 (9): 1724–35. PMC 1994707. PMID 17907809. doi:10.1371/journal.pgen.0030161.
- ↑ Pimentel H, Bray NL, Puente S, Melsted P, Pachter L (July 2017). "Differential analysis of RNA-seq incorporating quantification uncertainty". Nature Methods 14 (7): 687–690. PMID 28581496. doi:10.1038/nmeth.4324.
- ↑ Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L (January 2013). "Differential analysis of gene regulation at transcript resolution with RNA-seq" (PDF). Nature Biotechnology 31 (1): 46–53. PMC 3869392. PMID 23222703. doi:10.1038/nbt.2450.
- ↑ Frazee AC, Pertea G, Jaffe AE, Langmead B, Salzberg SL, Leek JT (March 2015). "Ballgown bridges the gap between transcriptome assembly and expression analysis". Nature Biotechnology 33 (3): 243–6. PMC 4792117. PMID 25748911. doi:10.1038/nbt.3172.
- ↑ 92,0 92,1 Sahraeian SM, Mohiyuddin M, Sebra R, Tilgner H, Afshar PT, Au KF, et al. (July 2017). "Gaining comprehensive biological insight into the transcriptome by performing a broad-spectrum RNA-seq analysis". Nature Communications 8 (1): 59. Bibcode:2017NatCo...8...59S. PMC 5498581. PMID 28680106. doi:10.1038/s41467-017-00050-4.
- ↑ Ziemann M, Eren Y, El-Osta A (August 2016). "Gene name errors are widespread in the scientific literature". Genome Biology 17 (1): 177. PMC 4994289. PMID 27552985. doi:10.1186/s13059-016-1044-7.
- ↑ Soneson C, Delorenzi M (March 2013). "A comparison of methods for differential expression analysis of RNA-seq data". BMC Bioinformatics 14: 91. PMC 3608160. PMID 23497356. doi:10.1186/1471-2105-14-91.
- ↑ Fonseca NA, Marioni J, Brazma A (30 September 2014). "RNA-Seq gene profiling--a systematic empirical comparison". PLOS ONE 9 (9): e107026. Bibcode:2014PLoSO...9j7026F. PMC 4182317. PMID 25268973. doi:10.1371/journal.pone.0107026.
- ↑ Seyednasrollah F, Laiho A, Elo LL (January 2015). "Comparison of software packages for detecting differential expression in RNA-seq studies". Briefings in Bioinformatics 16 (1): 59–70. PMC 4293378. PMID 24300110. doi:10.1093/bib/bbt086.
- ↑ Rapaport F, Khanin R, Liang Y, Pirun M, Krek A, Zumbo P, et al. (2013). "Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data". Genome Biology 14 (9): R95. PMC 4054597. PMID 24020486. doi:10.1186/gb-2013-14-9-r95.
- ↑ Conesa A, Madrigal P, Tarazona S, Gomez-Cabrero D, Cervera A, McPherson A, et al. (January 2016). "A survey of best practices for RNA-seq data analysis". Genome Biology 17 (1): 13. PMC 4728800. PMID 26813401. doi:10.1186/s13059-016-0881-8.
- ↑ Costa-Silva J, Domingues D, Lopes FM (21 December 2017). "RNA-Seq differential expression analysis: An extended review and a software tool". PLOS ONE 12 (12): e0190152. Bibcode:2017PLoSO..1290152C. PMC 5739479. PMID 29267363. doi:10.1371/journal.pone.0190152.
- ↑ 100,0 100,1 Keren H, Lev-Maor G, Ast G (May 2010). "Alternative splicing and evolution: diversification, exon definition and function". Nature Reviews. Genetics 11 (5): 345–55. PMID 20376054. doi:10.1038/nrg2776.
- ↑ Liu R, Loraine AE, Dickerson JA (December 2014). "Comparisons of computational methods for differential alternative splicing detection using RNA-seq in plant systems". BMC Bioinformatics 15 (1): 364. PMC 4271460. PMID 25511303. doi:10.1186/s12859-014-0364-4.
- ↑ Pachter, Lior (19 April 2011). "Models for transcript quantification from RNA-Seq" (en inglés). Bibcode:2011arXiv1104.3889P. arXiv:1104.3889.
- ↑ 103,0 103,1 Li YI, Knowles DA, Humphrey J, Barbeira AN, Dickinson SP, Im HK, Pritchard JK (January 2018). "Annotation-free quantification of RNA splicing using LeafCutter". Nature Genetics 50 (1): 151–158. PMC 5742080. PMID 29229983. doi:10.1038/s41588-017-0004-9.
- ↑ Anders S, Reyes A, Huber W (October 2012). "Detecting differential usage of exons from RNA-seq data". Genome Research 22 (10): 2008–17. PMC 3460195. PMID 22722343. doi:10.1101/gr.133744.111.
- ↑ Shen S, Park JW, Huang J, Dittmar KA, Lu ZX, Zhou Q, et al. (April 2012). "MATS: a Bayesian framework for flexible detection of differential alternative splicing from RNA-Seq data". Nucleic Acids Research 40 (8): e61. PMC 3333886. PMID 22266656. doi:10.1093/nar/gkr1291.
- ↑ Wang X, Cairns MJ (June 2014). "SeqGSEA: a Bioconductor package for gene set enrichment analysis of RNA-Seq data integrating differential expression and splicing". Bioinformatics 30 (12): 1777–9. PMID 24535097. doi:10.1093/bioinformatics/btu090.
- ↑ Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L (January 2013). "Differential analysis of gene regulation at transcript resolution with RNA-seq". Nature Biotechnology 31 (1): 46–53. PMC 3869392. PMID 23222703. doi:10.1038/nbt.2450.
- ↑ Hu Y, Huang Y, Du Y, Orellana CF, Singh D, Johnson AR, et al. (January 2013). "DiffSplice: the genome-wide detection of differential splicing events with RNA-seq". Nucleic Acids Research 41 (2): e39. PMC 3553996. PMID 23155066. doi:10.1093/nar/gks1026.
- ↑ Vaquero-Garcia J, Barrera A, Gazzara MR, González-Vallinas J, Lahens NF, Hogenesch JB, et al. (February 2016). "A new view of transcriptome complexity and regulation through the lens of local splicing variations". eLife 5: e11752. PMC 4801060. PMID 26829591. doi:10.7554/eLife.11752.
- ↑ Merino GA, Conesa A, Fernández EA (March 2019). "A benchmarking of workflows for detecting differential splicing and differential expression at isoform level in human RNA-seq studies". Briefings in Bioinformatics 20 (2): 471–481. PMID 29040385. doi:10.1093/bib/bbx122.
- ↑ 111,0 111,1 Marcotte EM, Pellegrini M, Thompson MJ, Yeates TO, Eisenberg D (November 1999). "A combined algorithm for genome-wide prediction of protein function". Nature 402 (6757): 83–6. Bibcode:1999Natur.402...83M. PMID 10573421. doi:10.1038/47048.
- ↑ 112,0 112,1 Giorgi FM, Del Fabbro C, Licausi F (March 2013). "Comparative study of RNA-seq- and microarray-derived coexpression networks in Arabidopsis thaliana". Bioinformatics 29 (6): 717–24. PMID 23376351. doi:10.1093/bioinformatics/btt053.
- ↑ Iancu OD, Kawane S, Bottomly D, Searles R, Hitzemann R, McWeeney S (June 2012). "Utilizing RNA-Seq data for de novo coexpression network inference". Bioinformatics 28 (12): 1592–7. PMC 3493127. PMID 22556371. doi:10.1093/bioinformatics/bts245.
- ↑ Eksi R, Li HD, Menon R, Wen Y, Omenn GS, Kretzler M, Guan Y (Nov 2013). "Systematically differentiating functions for alternatively spliced isoforms through integrating RNA-seq data". PLoS Computational Biology 9 (11): e1003314. Bibcode:2013PLSCB...9E3314E. PMC 3820534. PMID 24244129. doi:10.1371/journal.pcbi.1003314.
- ↑ Li HD, Menon R, Omenn GS, Guan Y (August 2014). "The emerging era of genomic data integration for analyzing splice isoform function". Trends in Genetics 30 (8): 340–7. PMC 4112133. PMID 24951248. doi:10.1016/j.tig.2014.05.005.
- ↑ Foroushani A, Agrahari R, Docking R, Chang L, Duns G, Hudoba M, et al. (March 2017). "Large-scale gene network analysis reveals the significance of extracellular matrix pathway and homeobox genes in acute myeloid leukemia: an introduction to the Pigengene package and its applications". BMC Medical Genomics 10 (1): 16. PMC 5353782. PMID 28298217. doi:10.1186/s12920-017-0253-6.
- ↑ Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. (August 2009). "The Sequence Alignment/Map format and SAMtools". Bioinformatics 25 (16): 2078–9. PMC 2723002. PMID 19505943. doi:10.1093/bioinformatics/btp352.
- ↑ DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. (May 2011). "A framework for variation discovery and genotyping using next-generation DNA sequencing data". Nature Genetics 43 (5): 491–8. PMC 3083463. PMID 21478889. doi:10.1038/ng.806.
- ↑ Battle A, Brown CD, Engelhardt BE, Montgomery SB (October 2017). "Genetic effects on gene expression across human tissues". Nature 550 (7675): 204–213. Bibcode:2017Natur.550..204A. PMC 5776756. PMID 29022597. doi:10.1038/nature24277.
- ↑ Richter F, Hoffman GE, Manheimer KB, Patel N, Sharp AJ, McKean D, et al. (March 2019). "ORE Identifies Extreme Expression Effects Enriched for Rare Variants". Bioinformatics. PMID 30903145. doi:10.1093/bioinformatics/btz202.
- ↑ Teixeira MR (December 2006). "Recurrent fusion oncogenes in carcinomas". Critical Reviews in Oncogenesis 12 (3–4): 257–71. PMID 17425505. doi:10.1615/critrevoncog.v12.i3-4.40.
- ↑ Weber AP (November 2015). "Discovering New Biology through Sequencing of RNA". Plant Physiology 169 (3): 1524–31. PMC 4634082. PMID 26353759. doi:10.1104/pp.15.01081.
- ↑ Bainbridge MN, Warren RL, Hirst M, Romanuik T, Zeng T, Go A, et al. (September 2006). "Analysis of the prostate cancer cell line LNCaP transcriptome using a sequencing-by-synthesis approach". BMC Genomics 7: 246. PMC 1592491. PMID 17010196. doi:10.1186/1471-2164-7-246.
- ↑ Cheung F, Haas BJ, Goldberg SM, May GD, Xiao Y, Town CD (October 2006). "Sequencing Medicago truncatula expressed sequenced tags using 454 Life Sciences technology". BMC Genomics 7: 272. PMC 1635983. PMID 17062153. doi:10.1186/1471-2164-7-272.
- ↑ Emrich SJ, Barbazuk WB, Li L, Schnable PS (January 2007). "Gene discovery and annotation using LCM-454 transcriptome sequencing". Genome Research 17 (1): 69–73. PMC 1716268. PMID 17095711. doi:10.1101/gr.5145806.
- ↑ Weber AP, Weber KL, Carr K, Wilkerson C, Ohlrogge JB (May 2007). "Sampling the Arabidopsis transcriptome with massively parallel pyrosequencing". Plant Physiology 144 (1): 32–42. PMC 1913805. PMID 17351049. doi:10.1104/pp.107.096677.
- ↑ Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M (June 2008). "The transcriptional landscape of the yeast genome defined by RNA sequencing". Science 320 (5881): 1344–9. Bibcode:2008Sci...320.1344N. PMC 2951732. PMID 18451266. doi:10.1126/science.1158441.
- ↑ "ENCODE Data Matrix". Consultado o 2013-07-28.
- ↑ "The Cancer Genome Atlas - Data Portal". Consultado o 2013-07-28.
Véxase tamén
[editar | editar a fonte]Outros artigos
[editar | editar a fonte]Ligazóns externas
[editar | editar a fonte]- RNA-Seq for Everyone: a high-level guide to designing and implementing an RNA-Seq experiment.
- Taguchi, Y.-h. (2019). "Comparative Transcriptomics Analysis". Encyclopedia of Bioinformatics and Computational Biology. pp. 814–818. ISBN 9780128114322. doi:10.1016/B978-0-12-809633-8.20163-5.
- Reference Module in Life Sciences